paired end sequencing r1 and r2
The amplicon was 550nt sequenced using 300nt x2 Illumina chemistry. The direction and positional order of the paired-end reads R1R2.

Mining For Structural Variations In Next Generation Sequencing Data Semantic Scholar
In R1 the short and long fragments exhibit similar position specific mismatch rate while in R2 the long fragments lead to much higher mismatch rate than short fragments which.
. Normally R1 and R2 are not reverse complements of each other. What are the best programsscripts. The map is 80 but the R1 R2 counts are.
R1139283692 R2137472495 This usually does not happen for PE sequencing which makes me a bit nervous to proceed with analysis. When you run cutadapt you give it the adapter sequence to trim and this is different for R1 and R2 reads. For each cluster that passes filter a single sequence is written to the corresponding samples R1 FASTQ file and for a paired-end run a single sequence is also.
The steps below can be. If the sequencing sample is the same as the original copy the read R1 should be mappable in forward direction in. The 2 reads should overlap in middle.
Heres what the options look like without running it on our files. Therefore a robust tool is needed to merge paired-end reads that exhibit varying overlap lengths because of varying target fragment lengths. Merging paired end reads R1 and R2 files 10-09-2013 0730 AM.
We present the PEAR. To anyone who may have dealt with Illumina MiSeq paired end reads. The reverse reads are sequenced in a reversed manner but the content of the reverse read is.
Paired-End Reads There are two FastQ files generated in an Illumina paired-end reads sequencing run. The files have this naming convention. For a Paired end reads you would have libraries that are constructed of fragments in both directions but they are never on the same bead and only one continuous read.
The diagrams show the paired-end reads R1 R2 derived from sequencing DNA fragments white boxes with sequencing adapters gray boxes on either end. Dear Galaxy experts I need to merge R1 and R2 Illumina paired end reads. First sequencing can either be single-end where each sample has only sequence data or paired-end where each sample has two sequence data R1 and R2.

Figure 1 Iguide Method For Crispr Off Target Detection Springerlink
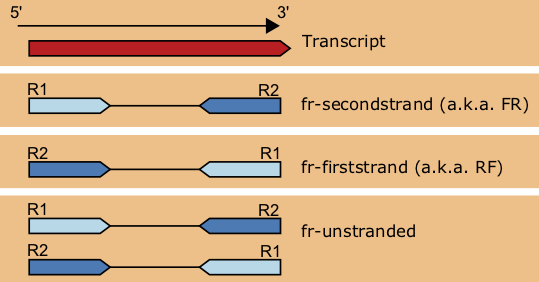
Chapter 6 Transcriptomics Applied Bioinformatics
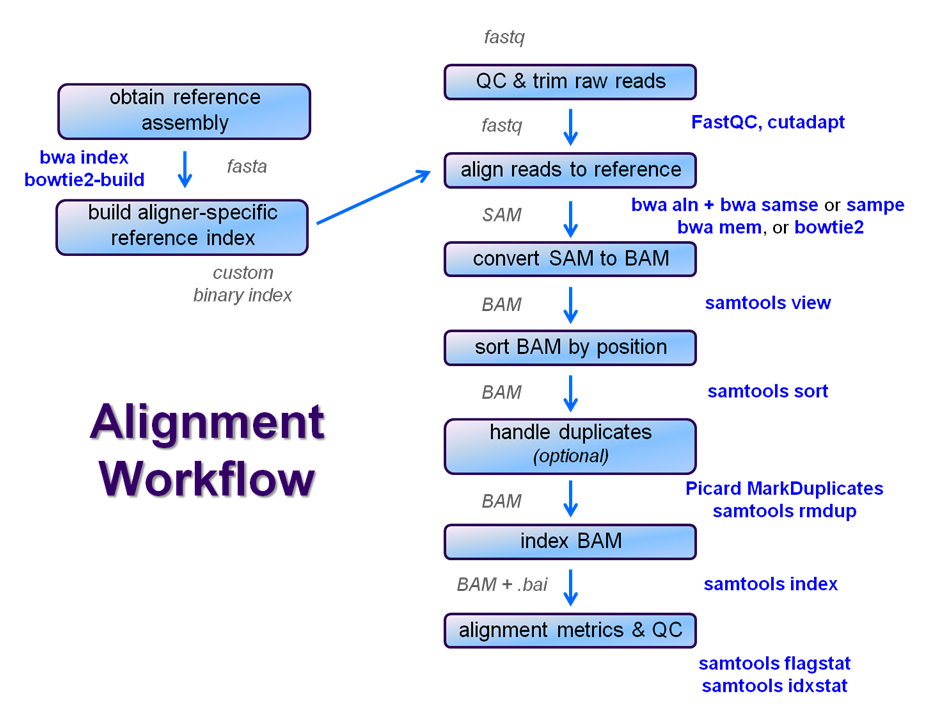
The Basic Alignment Workflow Core Ngs Tools Ut Austin Wikis
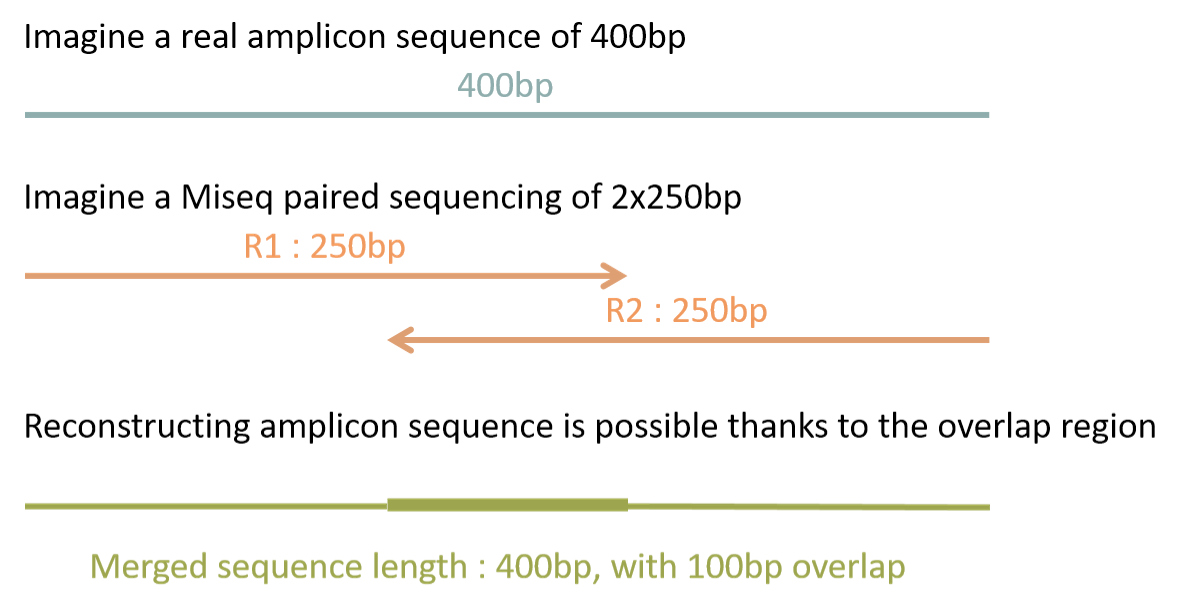
Frogs Faqs

Overview Of Pecc Seq A Library Preparation A Modified Pcr Free Download Scientific Diagram

Casava Files Of Paired End Reads General Discussion Qiime 2 Forum

Trouble With Merging Paired End Reads User Support Qiime 2 Forum
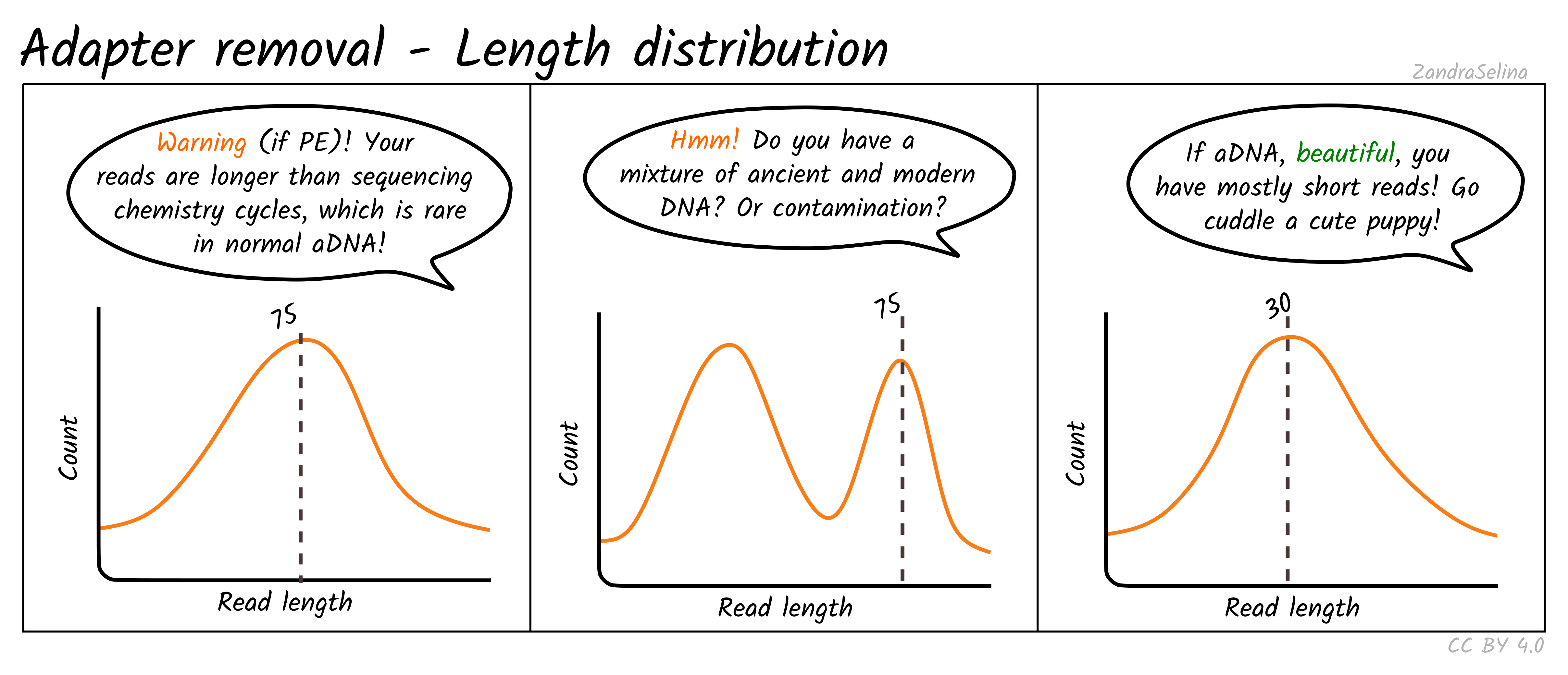
Eager Nf Core

Capturing Native Long Range Contiguity By In Situ Library Construction And Optical Sequencing Pnas
Im Tornado A Tool For Comparison Of 16s Reads From Paired End Libraries Plos One
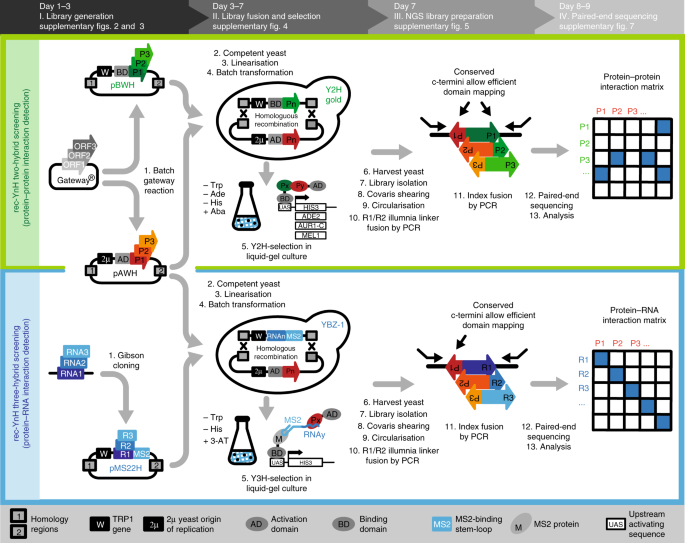
Rec Ynh Enables Simultaneous Many By Many Detection Of Direct Protein Protein And Protein Rna Interactions Nature Communications
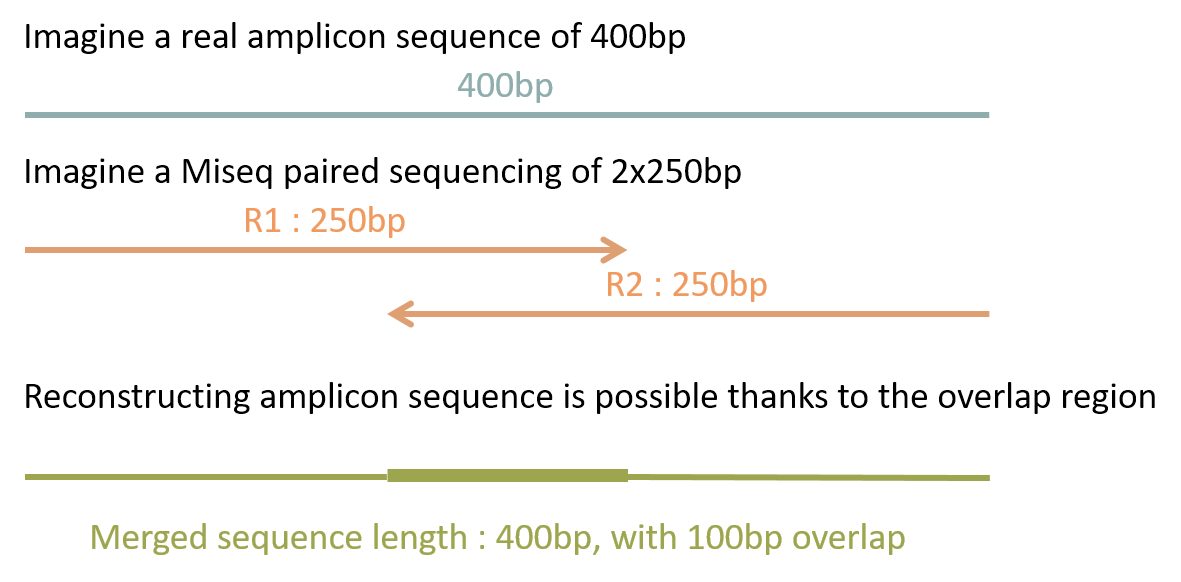
Galaxy

Comparison Of Aligned R1 R2 Reads Percentage Of Aligned Reads For The Download Scientific Diagram
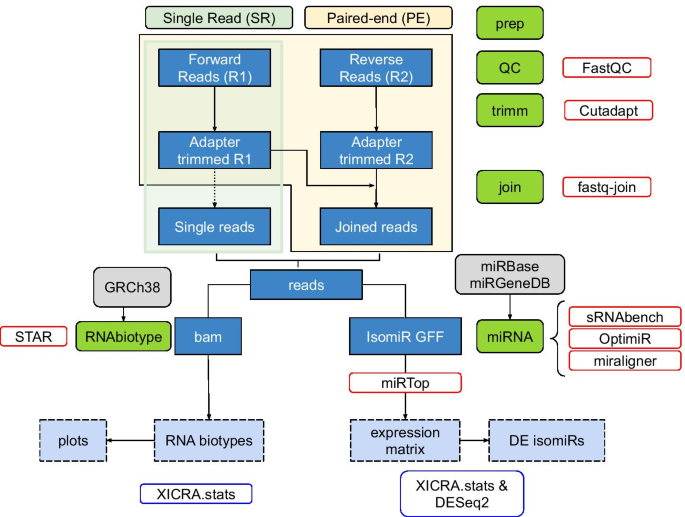
Paired End Small Rna Sequencing Reveals A Possible Overestimation In The Isomir Sequence Repertoire Previously Reported From Conventional Single Read Data Analysis Bmc Bioinformatics Full Text
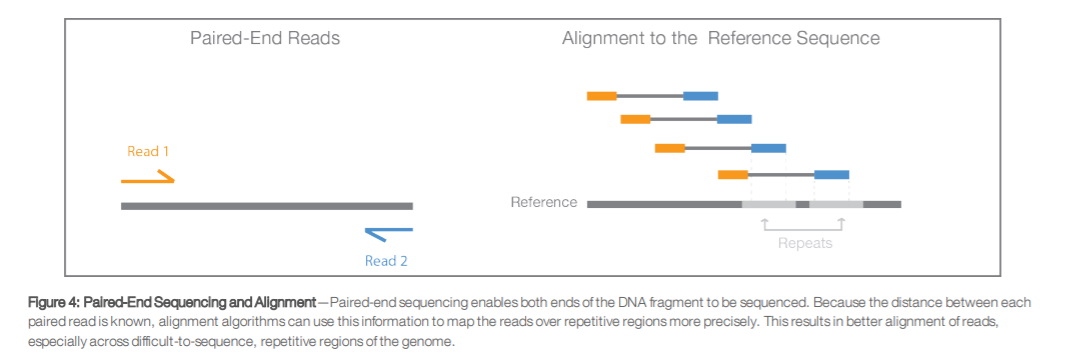
Illumina Sequencing Paired End Hc S Blog
Paired End Vs Single Read Sequencing Technology

Rna Seq How To Know Which Paired Reads Come From The Same Original Fragment

Paired End Sequencing The Inner Distance Between Paired Reads R1 And Download Scientific Diagram

Trouble With Merging Paired End Reads User Support Qiime 2 Forum